Траклир ДТ таб.дисперг. 32мг №56
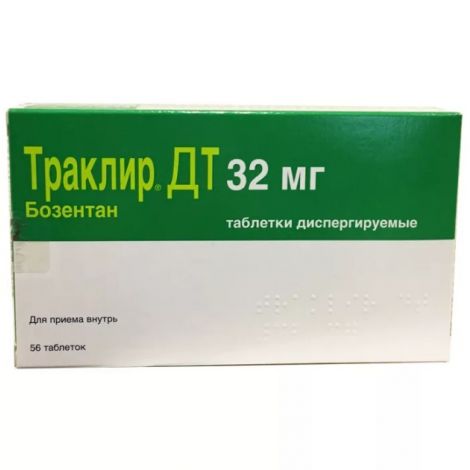
Внешний вид товара может отличаться от изображенного на фотографии
Описание
Условия хранения
Препарат следует хранить в недоступном для детей месте, при температуре не выше 25°C.
Способ применения и дозы
Диспергируемые таблетки следует принимать внутрь утром и вечером, независимо от времени приема пищи.
Подготовка к применению
Конструкция блистера, в котором размещены таблетки Траклир® ДТ, препятствует случайному доступу к ним детей.
Для того чтобы извлечь диспергируемую таблетку из блистера, необходимо:
1. По линии перфорации отделить от блистера одну ячейку, содержащую одну таблетку препарата.
2. Отогнуть края фольги в направлении стрелок и снять ее.
3. Выдавить таблетку из ячейки.
Для получения жидкой формы препарата Траклир®ДТ диспергируемую таблетку следует растворить в воде. Для этого в ложку наливают немного воды и кладут в нее таблетку. Таблетка должна быть целиком покрыта водой. При перемешивании добиваются полного растворения таблетки. Полученный раствор принимается пациентом. Для того, чтобы на ложке не осталось следов препарата, в нее добавляют еще немного воды и вновь проглатывают. Чтобы убедиться, что препарат принят полностью, нужно, при возможности, выпить еще стакан воды.
При необходимости диспергируемую таблетку можно разделить на части, разламывая вдоль нанесенных на нее рисок. Для этого таблетку рисками вверх зажимают с двух сторон от риски большим и указательным пальцами и разламывают на равные части.
Оставшаяся часть/части разделенной таблетки Траклир® ДТ может/могут храниться при Т от 15 °С до 25 °С , но должна/должны быть использована/ы в течение 7 дней.
Способ применения
Легочная артериальная гипертензия
Лечение и наблюдение должен проводить только врач, имеющий опыт лечения ЛАГ.
Применение у детей
Изучение параметров фармакокинетики бозентана у детей свидетельствует о том, что концентрация бозентана в плазме крови у детей с ЛАГ в возрасте от 1 года до 15 лет в среднем оказывается ниже, чем у взрослых пациентов, и не повышается с увеличением дозы препарата Траклир® ДГ более 2 мг/кг массы тела или частоты приема с 2-х до 3-х раз в день. Увеличение дозы препарата и кратности его приема, вероятно, не дают дополнительного клинического эффекта. На основании этих фармакокинетических данных, у детей с ЛАГ в возрасте 1 года и старше рекомендуется применять начальную и поддерживающую дозу 2 мг/кг массы тела утром и вечером.
При персистирующей ЛАГ новорожденных (ПЛГН) эффективность препарата Траклир® ДТ в стандартной схеме лечения не доказана.
Применение у взрослых
Применение диспергируемых таблеток изучено только у детей. Результаты сравнительной биодоступности таблеток, покрытых пленочной оболочкой, и диспергируемых таблеток у взрослых показали более низкую экспозицию бозентана при приеме диспергируемых таблеток (см. раздел "Фармакокинетика"). Таким образом, применение диспергируемых таблеток у взрослых может быть рекомендовано в тех случаях, когда пациенты не могут принимать таблетки, покрытые пленочной оболочкой.
У взрослых начальная доза составляет 62,5 мг 2 раза/день (утром и вечером) в течение 4 недель, затем дозу можно увеличить до поддерживающей дозы, которая составляет 125 мг 2 раза/день.
Этим рекомендациям нужно следовать и при возобновлении приема препарата Траклир® ДТ после перерыва в лечении.
Терапия в случае клинического ухудшения ЛАГ
Следует рассмотреть возможность применения альтернативных методов лечения в случае клинического ухудшения (например, при уменьшении дистанции ходьбы во время 6-минутного теста не менее чем на 10% по сравнению с исходными показателями), несмотря на применение препарата Траклир® ДТ в течение не менее 8 недель (из них в рекомендуемой дозе - не менее 4 недель). Однако у части пациентов при неэффективности препарата Траклир® ДТ после 8 недель применения, положительный эффект может наблюдаться после дополнительных 4-8 недель лечения. При наступлении клинического ухудшения спустя несколько месяцев лечения препаратом Траклир® ДТ, целесообразность его дальнейшего применения следует оценить заново. Увеличение дозы препарата Траклир® ДТ до 250 мг 2 раза/день у некоторых пациентов, при недостаточной эффективности его дозы 125 мг 2 раза/день, может способствовать некоторому повышению толерантности к физической нагрузке. Следует тщательно взвесить соотношение польза/риск для принятия решения об увеличении дозы препарата, принимая во внимание зависимость гепатотоксического действия препарата Траклир® ДТ от его дозы.
Действия в случае пропуска приема препарата
Если пациент забыл принять Траклир® ДТ, пропущенную дозу препарата следует принять как можно скорее, после чего лечение продолжают в обычном режиме. Не следует принимать одновременно 2 дозы препарата для восполнения пропущенной дозы.
Прекращение терапии
Имеется ограниченный опыт внезапного прекращения терапии препаратом Траклир® ДТ пациентов с ЛАГ, при этом данных о резком ухудшении ЛАГ не получено. Тем не менее, в целях предотвращения ухудшения состояния пациента из-за возможного развития синдрома "отмены, рекомендуется постепенное снижение дозы (наполовину в течение 3- 7 дней), на фоне альтернативной терапии. Рекомендуется регулярный контроль клинического состояния пациента в период отмены лечения.
Если принято решение об отмене препарата Траклир® ДТ, его дозу следует снижать постепенно, одновременно начиная альтернативную терапию.
Снижение числа новых дигитальных язв у взрослых с системной склеродермией
Назначение лечения и наблюдение за ним должен осуществлять только врач, имеющий опыт лечения системной склеродермии.
У взрослых пациентов начальная доза препарата Траклир® ДТ составляет 62,5 мг 2 раза/день в течение 4 недель, затем доза увеличивается до поддерживающей дозы - 125 мг 2 раза/день. Этим рекомендациям нужно следовать и при возобновлении приема препарата Траклир® ДТ после перерыва в лечении.
Необходимо проводить регулярное исследование клинического состояния пациента для решения вопроса о целесообразности продолжения лечения. Необходима тщательная оценка соотношения польза/риск, учитывая возможность гепатотоксического действия препарата Траклир® ДТ.
Клинический опыт применения препарата по данному показанию не превышает 6 месяцев.
Применение у детей
Данные об эффективности и безопасности применения препарата Траклир® ДТ по данному показанию у пациентов до 18 лет отсутствуют (см. раздел "Противопоказания"). Фармакокинетику препарата Траклир® ДТ у детей с данным заболеванием не изучали.
Применение в особых группах пациентов
Нарушение функции печени
Не требуется коррекции дозы для пациентов с печеночной недостаточностью легкой степени тяжести (класс А по классификации Чайлд-Пью, см. раздел "Фармакокинетика").
Траклир® ДТ противопоказан пациентам с печеночной недостаточностью умеренной и тяжелой степени тяжести (классы В и С по классификации Чайлд-Пью).
Нарушение функции почек
Для пациентов с нарушениями функции почек и пациентов, находящихся на гемодиализе, не требуется коррекции дозы препарата.
Применение у пожилых пациентов
У пациентов старше 65 лет не требуется коррекции дозы препарата.Противопоказания
- Повышенная чувствительность к бозентану или любому из компонентов препарата
- Печеночная недостаточность умеренной и тяжелой степени тяжести (классы В и С по классификации Чайлд-Пью)
- Исходное повышение активности «печеночных» трансаминаз: ACT (аспартатаминотрансферазы) и/или АЛТ (аланинаминотрансферазы) более чем в 3 раза от верхней границы нормы (ВГН)
- Тяжелая артериальная гипотензия (систолическое артериальное давление (АД) менее 85 мм рт.ст. у взрослых; систолическое АД < 80% нижней границы нормы, соответствующей возрасту и полу ребенка)
- Одновременный прием циклоспорина А
- Беременность
- Применение у женщин с сохраненным репродуктивным потенциалом, не пользующихся надежными методами контрацепции
- Период грудного вскармливания (отсутствуют клинические данные)
- Применение у детей до 18 лет для уменьшения числа новых дигитальных язв (в связи с отсутствием клинических данных)
- Персистирующая ЛАГ новорожденных (ПЛГН)
- Детский возраст до 3-х месяцев при ЛАГ
- Фенилкетонурия (препарат содержит аспартам).
С осторожностью:
- Артериальная гипотензия;
- Применение у детей в возрасте до 2-х лет (опыт клинического применения ограничен).
Беременность и лактация:
Беременность
В доклинических исследованиях установлена репродуктивная токсичность бозентана (тератогенное и эмбриотоксическое действие). Данных о применении препарата Траклир® ДТ у беременных не получено. Возможный риск для человека неизвестен. Применение препарата Траклир® ДТ во время беременности противопоказано.
Грудное вскармливание
Не установлено, проникает ли бозентан в грудное молоко. Если необходимо проведение терапии препаратом Траклир® ДТ в период лактации, грудное вскармливание рекомендуется прекратить.
Фертильность:
Применение у женщин с сохраненным репродуктивным потенциалом
Перед началом лечения препаратом Траклир® ДТ у женщин с сохраненным репродуктивным потенциалом следует подтвердить отсутствие беременности, обсудить надежные методы по предупреждению беременности и выбрать подходящие для пациента методы контрацепции. Пациенты и врачи должны учитывать тот факт, что бозентан может снижать эффективность гормональных контрацептивных средств вследствие фармакокинетического взаимодействия (см. раздел "Взаимодействие с другими лекарственными препаратами"). Поэтому женщинам с сохраненным репродуктивным потенциалом не рекомендуется использовать метод гормональной контрацепции (включая лекарственные препараты, применяемые внутрь, в виде инъекций, трансдермальных терапевтических систем или имплантатов) в качестве единственного метода; им необходимо применять дополнительный или альтернативный надежный метод контрацепции. При сомнениях в надежности выбранного метода контрацепции пациентке следует обратиться к врачу-гинекологу для индивидуального подбора метода контрацепции. Учитывая возможную неэффективность гормональной контрацепции и высокий риск негативного влияния беременности на течение ЛАГ, во время терапии препаратом Траклир® ДТ рекомендуется ежемесячно проводить тест на беременность, что позволит диагностировать беременность на ранних сроках.
Фертильность
В доклинических исследованиях у самцов выявили изменения в семенниках. Результаты клинического исследования показали, что у 8 из 24 пациентов-мужчин с ЛАГ через 3 или 6 месяцев лечения бозентаном отмечалось уменьшение концентрации спермы на 42% и более от исходных значений. На основании клинических и доклинических данных нельзя исключить риск негативного влияния бозентана на сперматогенез у мужчин. Нельзя исключить неблагоприятное влияние длительного лечения бозентаном на сперматогенез у пациентов-мальчиков.Меры предосторожности
- Артериальная гипотензия;
- применение у детей в возрасте до 2 лет (опыт клинического применения ограничен).Применение при беременности и в период лактации
В доклинических исследованиях установлена репродуктивная токсичность бозентана (тератогенное и эмбриотоксическое действие). Данных о применении препарата Траклир® ДТ у беременных не получено. Возможный риск для человека неизвестен. Применение препарата Траклир® ДТ при беременности противопоказано.
Перед началом лечения препаратом Траклир® ДТ у женщин репродуктивного возраста следует подтвердить отсутствие беременности, обсудить надежные методы по предупреждению беременности и выбрать подходящие для пациента методы контрацепции. Пациенты и врачи должны учитывать тот факт, что бозентан может снижать эффективность гормональных контрацептивных средств вследствие фармакокинетического взаимодействия. Поэтому женщинам репродуктивного возраста не рекомендуется использовать метод гормональной контрацепции (включая лекарственные препараты, применяемые внутрь, в виде инъекций, трансдермальных терапевтических систем или имплантатов) в качестве единственного метода; им необходимо применять дополнительный или альтернативный надежный метод контрацепции. Если имеются сомнения в отношении надежности выбранного метода контрацепции, пациентке следует обратиться к врачу-гинекологу для индивидуального подбора метода контрацепции. Учитывая возможную неэффективность гормональной контрацепции и высокий риск негативного влияния беременности на течение ЛАГ, во время терапии препаратом Траклир® ДТ рекомендуется ежемесячно проводить тест на беременность, что позволит диагностировать беременность на ранних сроках.
Не установлено, выделяется ли бозентан с грудным молоком. При необходимости проведения терапии препаратом Траклир® ДТ в период лактации грудное вскармливание рекомендуется прекратить.
Фертильность
В доклинических исследованиях у самцов выявили изменения в семенниках. Результаты клинического исследования показали, что у 8 из 24 пациентов-мужчин с ЛАГ через 3 или 6 месяцев лечения бозентаном отмечалось уменьшение концентрации спермы на 42% и более от исходных значений. На основании клинических и доклинических данных нельзя исключить риск негативного влияния бозентана на сперматогенез у мужчин. Нельзя исключить неблагоприятное влияние длительного лечения бозентаном на сперматогенез у пациентов-мальчиков.Показания к применению
Легочная артериальная гипертензия (ЛАГ) II-III функционального класса (ФК) по классификации ВОЗ с целью повышения толерантности к физическим нагрузкам и улучшения клинических симптомов, включая:
- первичную (идиопатическую и наследственную) легочную артериальную гипертензию;
- вторичную легочную артериальную гипертензию на фоне склеродермии при отсутствии значимого интерстициального поражения легких;
- легочную артериальную гипертензию, ассоциированную с врожденными пороками сердца и с нарушениями показателей гемодинамики по типу синдрома Эйзенменгера.
Снижение числа новых дигитальных язв у взрослых пациентов с системной склеродермией и дигитальными язвами (в случае, когда пациенты не могут принимать таблетки, покрытые пленочной оболочкой).Фармакокинетика
Фармакокинетические параметры у здоровых добровольцев зависят от дозы и времени приема бозентана. После приема внутрь системная экспозиция бозентана пропорциональна в дозах до 500 мг. При приеме внутрь бозентана в более высоких дозах Cmax в плазме крови и AUC увеличиваются менее пропорционально дозе. Ограниченные фармакокинетические данные свидетельствуют о том, что экспозиция бозентана у взрослых пациентов с ЛАГ примерно в 2 раза выше, чем у здоровых добровольцев.
Всасывание
После приема препарат внутрь Cmax в плазме крови достигается через 3-5 ч. Абсолютная биодоступность бозентана у здоровых добровольцев после приема внутрь составляет около 50% и не зависит от приема пищи.
Распределение
Бозентан в высокой степени (более 98%) связывается с белками плазмы крови, в основном, с альбумином. Бозентан не проникает в эритроциты. После однократного в/в введения в дозе 250 мг Vd составляет 18 л.
Метаболизм и выведение
После однократного в/в введения бозентана в дозе 250 мг его клиренс составляет 8.2 л/ч. T1/2 - 5.4 ч.
При многократном применении концентрация бозентана в плазме крови снижается постепенно и составляет 50-65% от концентрации при однократном применении. Равновесное состояние достигается в течение 3-5 дней.
Бозентан метаболизируется в печени при участии изоферментов цитохрома Р450 CYP2C9 и CYP3A4. Бозентан выводится через кишечник с желчью, менее 3% принятой дозы внутрь выводится почками.
В процессе метаболизма бозентана образуются 3 метаболита, один из которых обладает фармакологической активностью. Фармакологически активный метаболит преимущественно выводится с желчью. У взрослых пациентов концентрация активного метаболита в плазме крови выше, чем у здоровых добровольцев. У пациентов с признаками холестаза системное воздействие этого метаболита может возрастать.
Бозентан является индуктором изоферментов CYP2C9 и CYP3A4, а также, возможно, изофермента CYP2C19 и Р-гликопротеина. In vitro бозентан подавляет активность BSEP (помпа выведения солей желчных кислот).
В исследованиях in vitro показано, что бозентан не оказывает значимого ингибирующего эффекта на ряд изоферментов CYP (CYP1A2, 2А6, 2В6, 2С8, 2С9, 2D6, 2Е1, 3А4). Следовательно, бозентан не повышает концентрацию в плазме крови лекарственных средств, метаболизм которых опосредован данными изоферментами.
Сравнение лекарственных форм
В перекрестном исследовании изучали параметры фармакокинетики у 16 взрослых здоровых добровольцев, которые принимали бозентан в лекарственной форме таблетки, покрытые пленочной оболочкой, в дозе 62.5 мг, или в лекарственной форме диспергируемые таблетки в дозе 64 мг (2 таб. по 32 мг). После приема диспергируемых таблеток концентрация бозентана в плазме крови оказалась ниже, чем после приема таблеток, покрытых пленочной оболочкой (соотношение средних геометрических значений AUC0-∞ 0.87). Вид лекарственной формы не оказывал значимого влияния на Tmax и T1/2 бозентана.
Фармакокинетика у пациентов особых групп
На основании результатов исследований предполагается, что на фармакокинетику бозентана у взрослых пациентов не оказывают существенного влияния такие факторы, как пол, масса тела, расовая принадлежность или возраст.
Дети
Исследования фармакокинетики бозентана проводили также у детей. Тем не менее, фармакокинетические характеристики бозентана у детей до 2 лет полностью не определены ввиду ограниченных данных у этой категории пациентов.
Результаты исследования фармакокинетики у 19 детей с ЛАГ в возрасте от 3 до 15 лет при однократном и многократном приеме внутрь бозентана в лекарственной форме таблетки, покрытые оболочкой, в дозе 2 мг/кг массы тела 2 раза/сут, свидетельствуют о том, что экспозиция бозентана снижается со временем в полном соответствии с аутоиндукционными свойствами бозентана. Средние значения AUC (CV%) бозентана у детей, получающих бозентан в дозах 31.25 мг, 62.5 мг или 125 мг 2 раза/сут, составляли 3.496 (49%), 5.228 (79%) и 6.124 (27%) нг×ч/мл, соответственно, и были ниже среднего значения этого показателя (8.149 (47%) нг×ч/мл) у взрослых пациентов с ЛАГ, принимавших бозентан в дозе 125 мг 2 раза/сут. В равновесном состоянии системная экспозиция у детей с массой тела 10-20 кг, 20-40 кг и более 40 кг составила, соответственно, 43%, 67% и 75% от соответствующих показателей у взрослых.
Исследование диспергируемых таблеток у 36 пациентов с ЛАГ в возрасте от 2 до 11 лет не обнаружило пропорциональной зависимости фармакокинетических параметров от величины дозы, поскольку в равновесном состоянии концентрация бозентана в плазме крови и значения AUC оказались близки при приеме внутрь бозентана в дозе 2 мг/кг и 4 мг/кг массы тела 2 раза/сут соответственно (AUCtt: 3.577 и 3.371 нг×ч/мл). Общая системная экспозиция на фоне приема бозентана в дозе 125 мг 2 раза/сут у детей была примерно в 2 раза ниже, чем у взрослых пациентов, при этом в большинстве случаев показатели экспозиции у детей и взрослых совпадали.
В целом, в группе детей (n=31), принимавших бозентан в дозе 2 мг/кг массы тела 2 раза/сут, его средняя суточная экспозиция составляла 8.535 нг×ч/мл, a AUCtt - 4.268 нг×ч/мл (CV: 61%). У детей в возрасте от 3 месяцев до 2 лет средняя суточная экспозиция составила 7.879 нг×ч/мл, a AUCtt - 3.939 нг×ч/мл (CV: 72%). У детей от 3 месяцев до 1 года (n=2) - AUCtt составляла 5.914 нг×ч/мл (CV: 85%), а у пациентов от 1 года до 2 лет (n=7) AUCtt - 3.507 нг×ч/мл (CV: 70%). У пациентов старше 2 лет (n=22) средняя суточная экспозиция бозентана составила 8.820 нг×ч/мл, a AUCtt - 4.410 нг×ч/мл (CV: 58%). Применение бозентана в дозе 2 мг/кг 3 раза/сут не приводило к повышению концентрации бозентана в плазме крови, и средняя суточная экспозиция при этом составляла 7.275 нг×ч/мл (CV: 83%, n=27).
Полученные данные подтверждают, что концентрация бозентана в плазме крови достигает плато у детей при приеме в более низких дозах по сравнению со взрослыми. Кроме того, прием препарата в дозах выше 2 мг/кг 2 раза/сут (4 мг/кг 2 раза/сут или 2 мг/кг 3 раза/сут) не приводит к увеличению экспозиции бозентана у детей.
В исследовании, проведенном у новорожденных, концентрации бозентана повышались медленно и продолжали увеличиваться после окончания первого приема препарата, демонстрируя низкую экспозицию (AUC0-12 в цельной крови 164 нг×ч/мл, n=11). В равновесном состоянии AUCtt составила 6.165 нг×ч/мл (CV: 133%, n=7), что сопоставимо с величиной экспозиции у взрослых пациентов с ЛАГ при приеме бозентана в дозе 125 мг 2 раза/сут, учитывая соотношения распределения препарата в цельной крови и плазме крови, равное 0.6.
Значимость полученных данных в отношении гепатотоксичности препарата не определена. На фармакокинетику бозентана не оказывают существенного влияния пол и сопутствующее применение эпопростенола.
Нарушение функции печени
У пациентов с печеночной недостаточностью легкой степени (класс А по классификации Чайлд-Пью), существенных изменений показателей фармакокинетики препарата не отмечено. У этих пациентов равновесная AUC бозентана была на 9% выше, а его активного метаболита, Ro 48-5033, - на 33% выше по сравнению с аналогичным показателем у здоровых добровольцев.
Влияние печеночной недостаточности умеренной степени тяжести (класс В по классификации Чайлд-Пью) на фармакокинетические параметры бозентана и его основного метаболита, Ro 48-5033, изучали у 5 пациентов с ЛАГ, обусловленной портальной гипертензией, и печеночной недостаточностью умеренной степени тяжести, а также у 3 пациентов с ЛАГ, обусловленной другими причинами, и нормальной функцией печени. У пациентов с печеночной недостаточностью класса В среднее значение равновесной AUC бозентана составило 360 (212-613) нг×ч/мл, т.е. было в 4,7 раза выше, а среднее значение AUC активного метаболита Ro 48-5033 составило 106 (58.4-192) нг×ч/мл, т.е. в 12.4 раза выше, чем у пациентов с нормальной функцией печени (бозентан: среднее AUC: 76.1 [9.07-638] нг×ч/мл; Ro 48-5033: среднее AUC: 8.57 [1.28-57.2] нг×ч/мл). Несмотря на небольшое число пациентов и высокую вариабельность полученных данных, эти результаты указывают на значительное увеличение системной экспозиции бозентана и его основного метаболита Ro 48-5033 у пациентов с печеночной недостаточностью умеренной степени тяжести (класс В по классификации Чайлд-Пью).
Фармакокинетика бозентана у пациентов с тяжелой печеночной недостаточностью (класс С по классификации Чайлд-Пью) не изучена. Применение препарата Траклир®ДТ противопоказано у пациентов с печеночной недостаточностью умеренной и тяжелой степенями тяжести (класс В или С по классификации Чайлд-Пью).
Нарушение функции почек
У пациентов с почечной недостаточностью тяжелой степени (КК 15-30 мл/мин), концентрация бозентана в плазме крови снижается примерно на 10%. Концентрация метаболитов бозентана в плазме крови возрастает примерно в 2 раза по сравнению с пациентами с нормальной функцией почек. У пациентов с почечной недостаточностью не требуется коррекции дозы. Применение бозентана на фоне гемодиализа не изучали. Учитывая физико-химические свойства бозентана и его высокую степень связывания с белками плазмы крови, значительного выведения бозентана из сосудистого русла во время гемодиализа не ожидается.Фармакологическое действие
Фармакодинамика:
Бозентан является неселективным антагонистом эндотелиновых рецепторов типа ЕТA и ЕТв. Бозентан снижает как легочное, так и системное сосудистое сопротивление, приводя к повышению сердечного выброса без увеличения частоты сердечных сокращений (ЧСС).
Нейрогормон эндотелии-1 (ЕТ-1) является одним из самых мощных среди известных в настоящий момент вазоконстрикторов, который также обладает способностью стимулировать фиброз, клеточную пролиферацию, гипертрофию и ремоделирование, и также проявляет провоспалительную активность. Эти эффекты индуцируются при связывании эндотелина-1 (ЕТ-1) с рецепторами ЕТА и ЕТв, расположенными в эндотелии и клетках гладкой мускулатуры сосудов. Концентрации ЕТ-1 в тканях и плазме крови повышаются при некоторых сердечно-сосудистых заболеваниях и болезни соединительной ткани, в том числе при легочной артериальной гипертензии (ЛАГ), склеродермии, острой и хронической сердечной недостаточности, ишемии миокарда, системной гипертензии и атеросклерозе, что предполагает участие ЕТ-1 в патогенезе этих заболеваний. При ЛАГ и сердечной недостаточности, в отсутствии антагонизма рецепторов к ЕТ, повышенные концентрации ЕТ-1 строго коррелируют с тяжестью и прогнозом указанных заболеваний.
Бозентан конкурирует с ЕТ-1 и другими ЕТ пептидами за связывание с ЕТA и ЕТв, с незначительно более высоким сродством к рецепторам ЕТA (Ki = 4,1-43 нмоль), по сравнению с рецепторами ЕТв (Ki = 38-730 нмоль).
Бозентан специфически блокирует ЕТ рецепторы и не связывается с другими рецепторами.
При изучении ЛАГ на моделях животных показано, что длительное введение бозентана внутрь снижает легочное сосудистое сопротивление и способствует обратному развитию гипертрофии сосудов легких и правого желудочка. Показано, что при легочном фиброзе бозентан уменьшает накопление коллагена в легких.
Результаты инвазивных исследований гемодинамики показали, что лечение бозентаном приводит к значительному повышению сердечного индекса, а также существенному снижению давления в легочной артерии, легочного сосудистого сопротивления и среднего давления в правом предсердии.
Длительное (в течение 12 и 16 недель) лечение взрослых пациентов с ЛАГ (первичной и вторичной, преимущественно ассоциированное со склеродермией) III-IV функционального класса (ФК) по классификации Всемирной организации здравоохранения (ВОЗ) бозентаном в комбинации с антикоагулянтами, вазодилататорами (блокаторами кальциевых каналов), диуретиками, кислородом и дигоксином, но не эпопростенолом, сопровождалось уменьшением выраженности симптомов ЛАГ и значительным увеличением толерантности к физическим нагрузкам (по результатам теста с 6-минутной ходьбой). Эти эффекты отмечались через 4 недели, отчетливо проявлялись через 8 недель и сохранялись до 28 недель в подгруппе пациентов активного лечения.
В исследовании пациентов с ЛАГ II ФК показано значительное увеличение времени до начала клинического ухудшения (комбинированная точка, включающая прогрессирование симптомов заболевания, госпитализацию вследствие ЛАГ и случаи смерти).
У пациентов с ЛАГ III ФК и пороками сердца в сочетании с нарушениями показателей гемодинамики по типу синдрома Эйзенменгера, увеличение среднего значения насыщения крови кислородом свидетельствовало о том, что бозентан не усугубляет гипоксемию, и что среднее значение легочного сосудистого сопротивления значительно снижается в группе бозентана.
При изучении бозентана у пациентов с ЛАГ III ФК в сочетании с ВИЧ-инфекцией показано повышение толерантности к физическим нагрузкам в сравнении с исходными данными.
В двух основных плацебо-контролируемых исследованиях и их открытых продлениях у всех пациентов, получающих бозентан, в течение длительного времени проводили оценку жизненно важных показателей. Средняя продолжительность приема бозентана составила 1,9 ± 0,7 лет (от 0,1 до 3,3 лет), клиническое состояние пациентов контролировали в среднем в течение 0,2 ± 0,6 лет. У большей части пациентов был подтвержден диагноз первичной ЛАГ (72%) и определен III ФК (84%) по классификации ВОЗ. Показатель выживаемости в группе в целом (согласно оценке по методу Каплан-Майера) через 1 год лечения бозентаном составил 93%, а через 2 года - 84%. У пациентов с системной склеродермией показатели выживаемости по методу Каплан-Майера были ниже.
Исследования, проведенные у детей с ЛАГ
Исследование параметров фармакокинетики бозентана проводили у детей с ЛАГ II-III ФК в возрасте от 3 до 15 лет в течение 12 недель терапии препаратом. Анализ гемодинамических параметров указывает на увеличение сердечного индекса (СИ) на 0,5 л/мин/м2, а также на умеренное снижение среднего давления в легочной артерии (ДЛА) до 8 мм рт.ст. и легочного сосудистого сопротивления (ЛСС) - до 389 дин-с\см5.
Бозентан, в лекарственной форме диспергируемые таблетки, применяли в дозе 2 и 4 мг/кг массы тела 2 раза в день у 36 пациентов в возрасте от 2 до 11 лет. Риск ухудшения течения заболевания (смерть, пересадка легких или госпитализация в связи с ухудшением ЛАГ), который оценивали по методу Каплан-Майера, через 2 года составил 78,9%. Показатель общей выживаемости, согласно оценке по Каплан-Майеру, через 2 года составил 91,2%.
В исследовании с участием 64 детей в возрасте от 3 месяцев до 11 лет со стабильной ЛАГ, которые получали бозентан в дозе 2 мг/кг массы тела 2 или 3 раза в день в течение 24 недель, установлено, что клиническое состояние большей части пациентов оставалось стабильным согласно функциональному классу (97% - при приеме препарата 2 раза в день, 100% - 3 раза в день) и результатам общей клинической оценки исследователей (94% - при приеме препарата 2 раза в день, 93% - 3 раза в день). Неосложненное течение ЛАГ согласно оценке по методу Каплан-Майера (случаи смерти, трансплантации легкого или госпитализации в связи с ухудшением ЛАГ), отмечали у 96,9% и 96,7% пациентов, принимающих препарат 2 и 3 раза в день, соответственно.
Клинически значимого преимущества применения препарата в дозе 2 мг/кг массы тела 3 раза в день по сравнению с его приемом 2 раза в день не установлено.
Исследовании, проведенные у новорожденных с персистирующей легочной гипертензией новорожденных (ПЛГН)
В двойном-слепом плацебо-контролируемом рандомизированном исследовании у детей, рожденных преждевременно или в нормальные сроки (гестационный возраст составил 36-42 недели) с ПЛГН и при субоптимальном ответе на ингаляции окиси азота (iNO), продолжительностью не менее 4 часов, в качестве дополнительного средства через назогастральный зонд вводили бозентан в лекарственной форме диспергируемые таблетки в дозе 2 мг/кг массы тела 2 раза в день (N=13) или плацебо (N=8) в момент максимальной концентрации iNO в плазме крови. Бозентан применяли до полной отмены iNO или до исчезновения эффекта от проводимого лечения (потребность в экстракорпоральной мембранной оксигенации [ЭКМО] или в применении альтернативного средства вазодилатации сосудов легких). Максимальная продолжительность лечения составила 14 дней.
Средняя продолжительность лечения составила 4,5 дн (от 0,5 до 10,0 дн) в группе бозентана и 4,0 дня (от 2,5 до 6,5 дн) - в группе плацебо.
Результаты, полученные в данной популяции, не указывают на дополнительные преимущества применения бозентана:- Средняя продолжительность применения iNO при одновременном применении бозентана составляла 3,7 дней и 2,9 дней - плацебо (р=0,34).
- Средняя продолжительность пребывания на ИВЛ составляла 10,8 дней при одновременном применении бозентана и 8,6 дней - плацебо (р=0,24).
- У одного пациента, получавшего бозентан эффект от лечения отсутствовал (потребность в ЭКМО, согласно требованиям протокола), что было диагностировано на основании повышения индекса оксигенации через 8 часов после введения первой дозы препарата. Состояние пациента улучшилось в течение 60 дней последующего наблюдения.
Совместное применение с эпопростенолом
Совместное применение бозентана и эпопростенола изучали в двух исследованиях: 10 из 19 пациентов детского возраста получали одновременно бозентан и эпопростенол в течение 12 недель. Профиль безопасности комбинированного лечения не отличался от профиля безопасности при применении препаратов по отдельности, переносимость комбинированного лечения у детей и взрослых пациентов была хорошая. Клинические преимущества указанного комбинированного лечения не наблюдались.
Системная склеродермия с язвенным поражением конечностей
Результаты двух клинических исследований у взрослых пациентов с системной склеродермией и язвенным поражением конечностей (в стадии обострения или в случаях, когда язвенное поражение отмечали в течение последнего года) показали, что в течение всего периода применения бозентана наблюдается достоверное уменьшение числа новых язвенных поражений конечностей по сравнению с плацебо.
У пациентов, получавших бозентан или плацебо в течение 16 недель, в среднем, отмечено 1,4 и 2,7 новых язвенных поражения, соответственно (р=0,0042). В исследовании продолжительностью 24 недели число новых язвенных поражений конечностей у пациента составляло, в среднем, 1,9 и 2,7 соответственно (р=0,0351). Влияние бозентана на скорость заживления язвенных поражений не установлено.
Фармакокинетика:
Фармакокинетические параметры у здоровых добровольцев зависят от дозы и времени приема бозентана. После приема внутрь системная экспозиция бозентана пропорциональна в дозах до 500 мг. При приеме внутрь более высоких доз бозентана максимальная концентрация в плазме крови (Сmах) и AUC (площадь под кривой "концентрация-время") увеличиваются менее пропорционально дозе. Ограниченные фармакокинетические данные свидетельствуют о том, что экспозиция бозентана у взрослых пациентов с ЛАГ примерно в 2 раза выше, чем у здоровых добровольцев.
Всасывание
Абсолютная биодоступность бозентана после приема внутрь составляет около 50% и не зависит от времени приема пищи. Сmах в плазме крови достигается через 3-5 ч после приема препарата внутрь.
Распределение
Бозентан в высокой степени (более 98%) связывается с белками плазмы крови, в основном, с альбумином. Бозентан не проникает в эритроциты. После однократного внутривенного введения в дозе 250 мг объем распределения составляет около 18 л.
Метаболизм и выведение
После однократного внутривенного введения бозентана в дозе 250 мг клиренс его составляет 8,2 л/ч, период полувыведения (Т1/2) - 5,4 ч. При многократном применении концентрация бозентана в плазме крови снижается постепенно до 50-65% от значений концентрации при однократном применении. Состояние устойчивого равновесия достигается в течение 3-5 дней.
Бозентан метаболизируется в печени при участии изоферментов цитохрома Р450 CYP2C9 и CYP3A4. Бозентан выводится через кишечник с желчью, менее 3% принятой дозы внутрь выводится почками.
Бозентан образует 3 метаболита, один из которых обладает фармакологической активностью. Фармакологически активный метаболит выводится преимущественно в неизменном виде с желчью. У взрослых пациентов концентрация активного метаболита в плазме крови выше, чем у здоровых добровольцев. У пациентов с признаками холестаза системное воздействие этого метаболита может возрастать.
Бозентан является индуктором изоферментов CYP2C9 и CYP3A4, а также возможно изофермента CYP2C19 и Р-гликопротеина. In vitro бозентан подавляет активность BSEP (помпа выведения солей желчных кислот).
В исследованиях in vitro показано, что бозентан не оказывает значимого ингибирующего эффекта на ряд изоферментов (CYP1A2, 2А6, 2В6, 2С8, 2С9, 2D6, 2Е1, 3А4). Следовательно, бозентан не повышает концентрацию в плазме крови лекарственных средств, метаболизм которых опосредован данными изоферментами.
Сравнение лекарственных форм
В перекрестном исследовании изучали параметры фармакокинетики у 16 взрослых здоровых добровольцев, которые принимали бозентан в лекарственной форме таблетки, покрытые пленочной оболочкой, в дозе 62,5 мг, или в лекарственной форме диспергируемые таблетки в дозе 64 мг (32 мг х 2). После приема диспергируемых таблеток концентрация бозентана в плазме крови оказалась ниже, чем после приема таблеток, покрытых пленочной оболочкой (соотношение средних геометрических значений AUC0-∞ 0,87). Вид лекарственной формы не оказывал значимого влияния на время достижения максимальной концентрации и Т1/2 бозентана.
Фармакокинетика в особых группах пациентов
Результаты исследований предполагают, что на фармакокинетику бозентана у взрослых пациентов не оказывают существенного влияния такие факторы, как пол, масса тела, расовая принадлежность или возраст.
Дети
Исследования фармакокинетики бозентана проводили также у детей. Тем не менее, фармакокинетические характеристики бозентана у детей до 2 лет полностью не определены ввиду ограниченных данных у этой категории пациентов.
Результаты исследования фармакокинетики у 19 детей с ЛАГ в возрасте от 3 до 15 лет при однократном и многократном приеме внутрь бозентана в лекарственной форме таблетки, покрытые оболочкой, в дозе 2 мг/кг массы тела 2 раза в день, свидетельствуют о том, что экспозиция бозентана снижается со временем в полном соответствии с автоиндукционными свойствами бозентана. Средние значения AUC (CV%) бозентана у детей, получающих бозентан в дозах 31,25 мг, 62,5 мг или 125 мг 2 раза в день, составляли 3,496 (49%), 5,228 (79%), и 6,124 (27%) нг*ч/мл, соответственно, и были ниже среднего значения этого показателя (8,149 (47%) нг*ч/мл) у взрослых пациентов с ЛАГ, принимавших бозентан в дозе 125 мг 2 раза в день. В равновесном состоянии системная экспозиция у детей с массой тела 10-20 кг, 20-40 кг и более 40 кг составила, соответственно, 43%, 67% и 75%, от соответствующих показателей у взрослых.
Исследование диспергируемых таблеток у 36 пациентов с ЛАГ в возрасте от 2 до 11 лет не обнаружило пропорциональной зависимости фармакокинетических параметров от величины дозы, поскольку в равновесном состоянии концентрация бозентана в плазме крови и значения AUC оказались близки при приеме внутрь бозентана в дозе 2 мг/кг и 4 мг/кг массы тела 2 раза в день, соответственно (AUCτ: 3,577 и 3,371 нг*ч/мл). Общая системная экспозиция на фоне приема бозентана в дозе 125 мг 2 раза в день у детей была примерно в 2 раза ниже, чем у взрослых пациентов, при этом в большинстве случаев показатели экспозиции у детей и взрослых совпадали.
В целом, в группе детей (n=31), принимавших бозентан в дозе 2 мг/кг массы тела 2 раза в день, его средняя суточная экспозиция составляла 8,535 нг*ч/мл, a AUCτ - 4,268 нг*ч/мл (CV: 61%). У детей в возрасте от 3 месяцев до 2 лет средняя суточная экспозиция составила 7,879 нг*ч/мл, a AUCτ - 3,939 нг*ч/мл (CV: 72%). У детей от 3 месяцев до 1 года (n=2) - AUCτ составляла 5,914 нг*ч/мл (CV: 85%), а у пациентов от 1 года до 2-х лет (n=7) AUCt - 3,507 нг*ч/мл (CV: 70%). У пациентов старше 2-х лет (n=22) средняя суточная экспозиция бозентана составила 8,820 нг*ч/мл, a AUCτ - 4,410 нг*ч/мл (CV: 58%). Применение бозентана в дозе 2 мг/кг 3 раза в день не приводило к повышению концентрации бозентана в плазме крови, и средняя суточная экспозиция при этом составляла 7,275 нг*ч/мл (CV: 83%, n=27).
Полученные данные подтверждают, что концентрация бозентана в плазме крови достигает плато у детей при приеме более низких доз по сравнению со взрослыми. Кроме того, прием препарата в дозах выше 2 мг/кг 2 раза в день (4 мг/кг 2 раза в день или 2 мг/кг 3 раза в день) не приводит к увеличению экспозиции бозентана у детей.
В исследовании, проведенном у новорожденных, концентрации бозентана повышались медленно и продолжали увеличиваться после окончания первого приема препарата, демонстрируя низкую экспозицию (AUC0-12 в цельной крови 164 нг*ч/мл, n=11). В равновесном состоянии AUCτ составила 6,165 нг*ч/мл (CV: 133%, n=7), что сопоставимо с величиной экспозиции у взрослых пациентов с ЛАГ при приеме бозентана в дозе 125 мг 2 раза в день, учитывая соотношения распределения препарата в цельной крови и плазме крови, равное 0,6.
Значимость полученных данных в отношении гепатотоксичности препарата не определена. На фармакокинетику бозентана не оказывают существенного влияния пол и сопутствующее применение эпопростенола.
Нарушение функции печени
У пациентов с печеночной недостаточностью легкой степени (класс А по классификации Чайлд-Пью), существенных изменений показателей фармакокинетики препарата не отмечено. У этих пациентов равновесная AUC бозентана была на 9% выше, а его активного метаболита, Ro 48-5033, - на 33% выше по сравнению с аналогичным показателем здоровых добровольцев.
Влияние печеночной недостаточности умеренной степени тяжести (класс В по классификации Чайлд-Пью) на фармакокинетические параметры бозентана и его основного метаболита, Ro 48-5033, изучали у 5 пациентов с ЛАГ, обусловленной портальной гипертензией, и печеночной недостаточностью умеренной степени тяжести, а также у 3 пациентов с ЛАГ, обусловленной другими причинами, и нормальной функцией печени. У пациентов с печеночной недостаточностью класса В среднее значение равновесной AUC бозентана составило 360 (212-613) нг*ч/мл, т.е. было в 4,7 раза выше, а среднее значение AUC активного метаболита Ro 48-5033 составило 106 (58,4-192) нг*ч/мл, т.е. в 12,4 раза выше, чем у пациентов с нормальной функцией печени (бозентан: среднее AUC: 76,1 [9,07-638] нг*ч/мл; Ro 48-5033: среднее AUC: 8,57 [1,28-57,2] нг*ч/мл). Несмотря на небольшое число пациентов и высокую вариабельность полученных данных, эти результаты указывают на значительное увеличение системной экспозиции бозентана и его основного метаболита Ro 48-5033 у пациентов с печеночной недостаточностью умеренной степени тяжести (класс В по классификации Чайлд-Пью).
Фармакокинетика бозентана у пациентов с тяжелой печеночной недостаточностью (класс С по классификации Чайлд-Пью) не изучена. Применение препарата Траклир®ДТ противопоказано у пациентов с печеночной недостаточностью умеренной и тяжелой степенями тяжести (класс В или С по классификации Чайлд-Пью).
Нарушение функции почек
У пациентов с почечной недостаточностью тяжелой степени тяжести (клиренс креатинина, КК, 15-30 мл/мин), концентрация бозентана в плазме крови снижается примерно на 10%. Концентрация метаболитов бозентана в плазме крови возрастает примерно в 2 раза по сравнению с пациентами с нормальной функцией почек. У пациентов с почечной недостаточностью не требуется коррекции дозы. Применение бозентана на фоне гемодиализа не изучали. Учитывая физико-химические свойства бозентана и его высокую степень связывания с белками плазмы крови, значительного выведения бозентана из сосудистого русла во время гемодиализа не ожидается.Описание товара
Плоскоцилиндрические таблетки от почти белого до бледно-желтого цвета, с фаской и двумя рисками, переходящими на боковую поверхность, на одной стороне и гравировкой "32" на другой стороне
Срок годности
3 года.
Форма выпуска
14 шт. - блистеры (4) - пачки картонные.